Research
My research interests are in the field of bioinformatics and computational biology, a highly interdisciplinary field that combines computer science, biology, mathematics, and statistics. Research in bioinformatics uses and develops new computational methods and models for processing, analyzing, and understanding biological data sets. Extracting knowledge from biological data has become a very complex task that often requires computational analysis as well as experimental biological analysis. My current research focuses on developing algorithms and tools necessary for gaining insight into the underlying genomic structure of a population.Computational tools for genetic reference populations
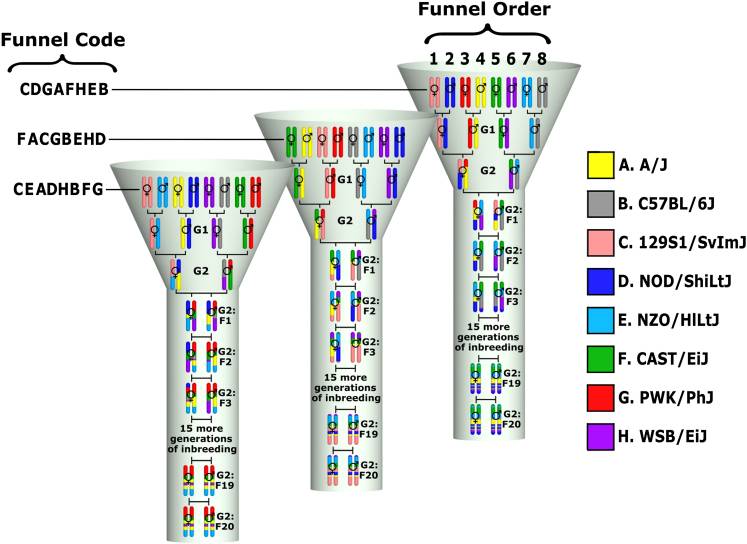
Genetic reference populations are defined as sets of individuals with fixed and known genomes that can be replicated infinitely. My work with genetic reference populations mainly focuses on the Collaborative Cross (CC), an inbred mouse population derived from eight inbred founder strains. The CC was designed to be a common platform for mammalian complex traits genetics that overcomes the limitations of existing resources and allows for systems genetics research. For researchers to best utilize this resource, they must first be able to map each genomic location of a CC mouse back to the founder from which it was inherited. I have developed tools and algorithms that assist with this mapping for various platforms, including genotyping arrays as well as high-throughput DNA sequencing data.
Mitochondrial Variant Detection and Analysis
My most recent work attempts to elucidate the functional significance of mitochondrial variants within human populations, particularly cancer patients and survivors. Mitochondrial variants occur frequently, both inherited and acquired, and have been implicated in a number of mitochondrial disorders, as well as cancer and aging. To understand the role that mitochondrial mutations have within a given population such as pediatric cancer, it is important to accurately detect these mutations and analyze their frequency within populations. I have developed various bioinformatics pipelines and tools that detect variants in mitochondrial DNA (mtDNA) from high-throughput sequencing data. For instance, a Rhodes undergraduate, Shane Elder, and myself designed MitoMut, a tool for detecting mtDNA deletions. I have also worked to understand the potential impact of these variants and looked at the correlation between these variants and traits of interest.